
")















Amsterdam Modeling Suite - AMS
ADF, BAND, DFTB, ReaxFF and COSMO-RS
Neue Version:
AMS 2025
Details zu den Neuerungen
SCM produces a suite of software to model chemical and physical properties. The density functional theory programs are accurate and efficient and the approximate quantum-based codes offer fast insight in complex systems. The programs work, in parallel, out of the box on any popular system (Windows, Mac, Linux/UNIX). Jobs for all programs may be prepared, executed and analyzed via a uniquely integrated user-friendly Graphical User Interface (GUI).
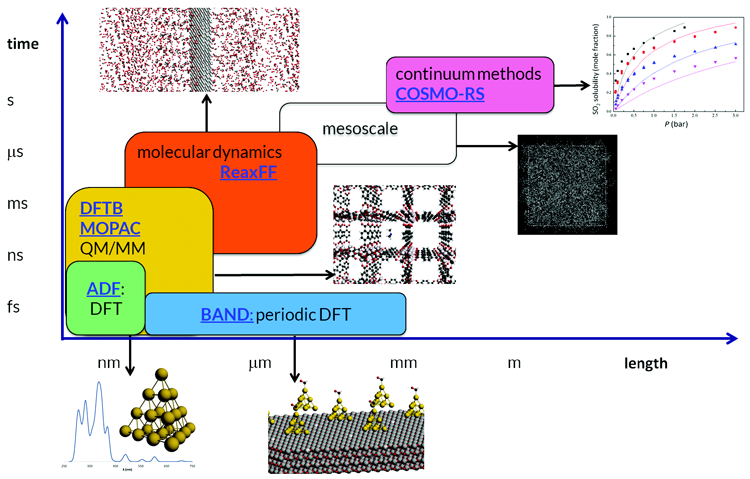
Overview over the Amsterdam Modeling Suite
Click to enlarge and browse the image gallery
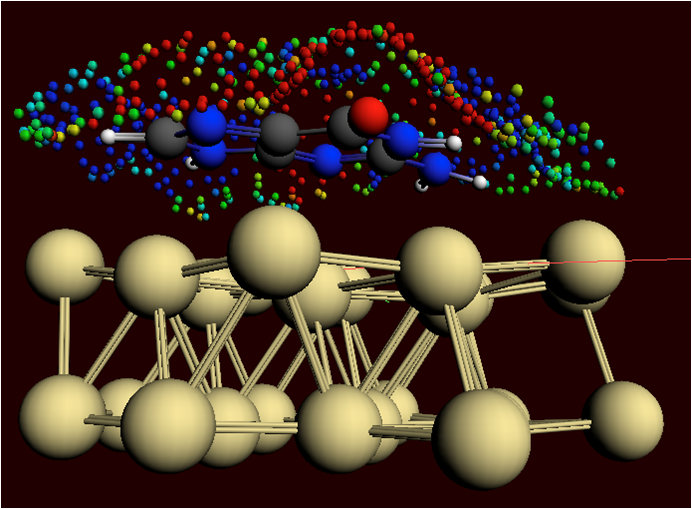
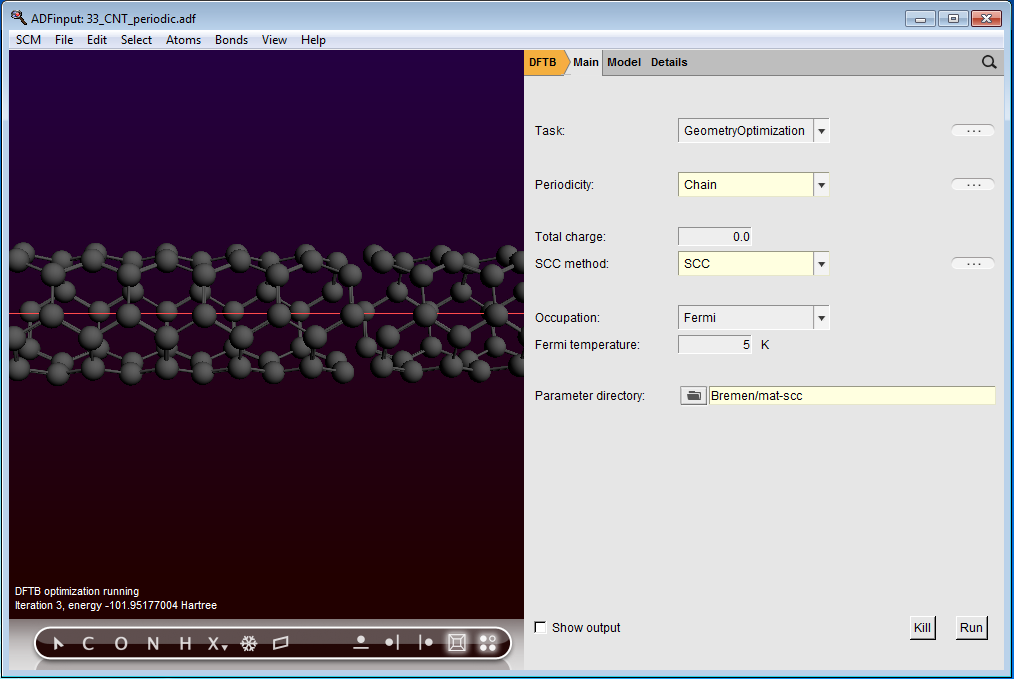
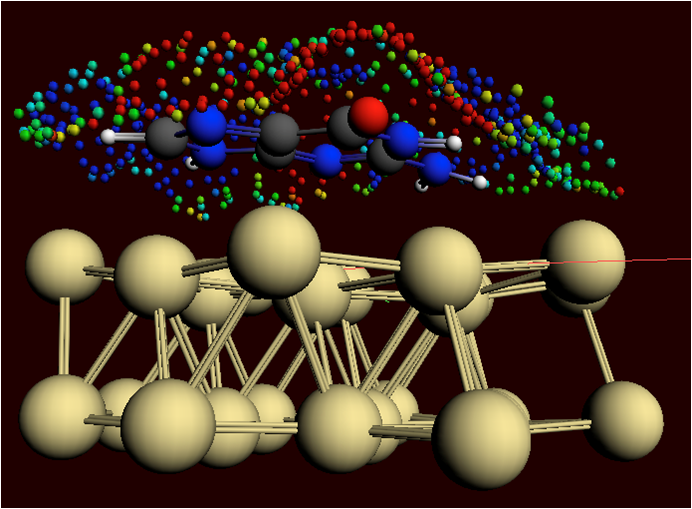
The ADF Program for molecular modeling
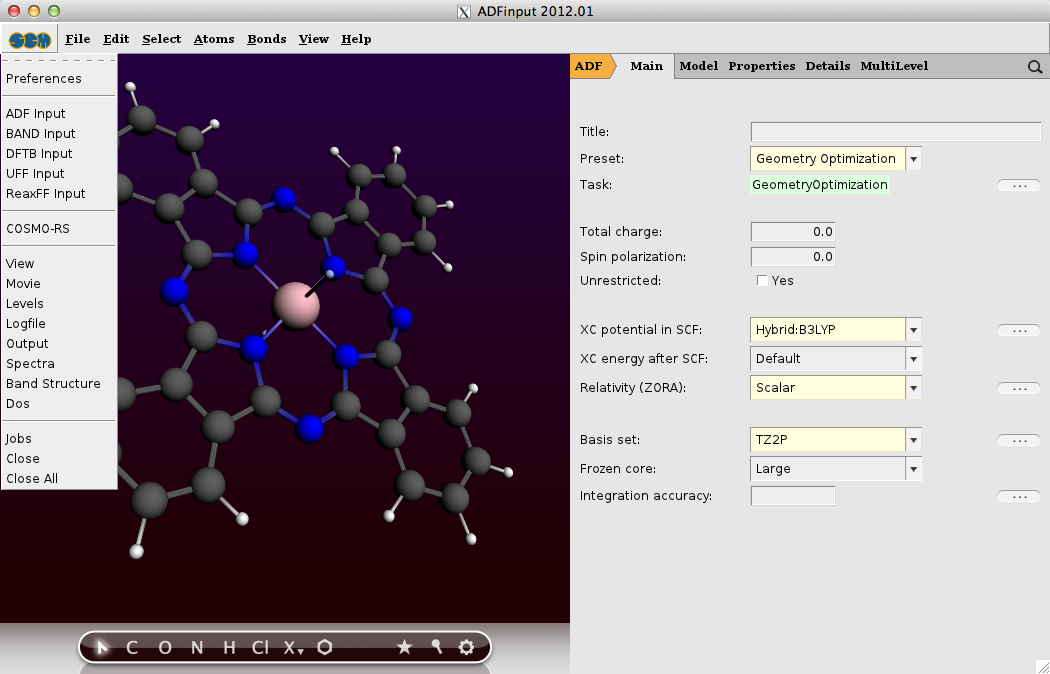
A calculation of a metal complex set-up with the GUI
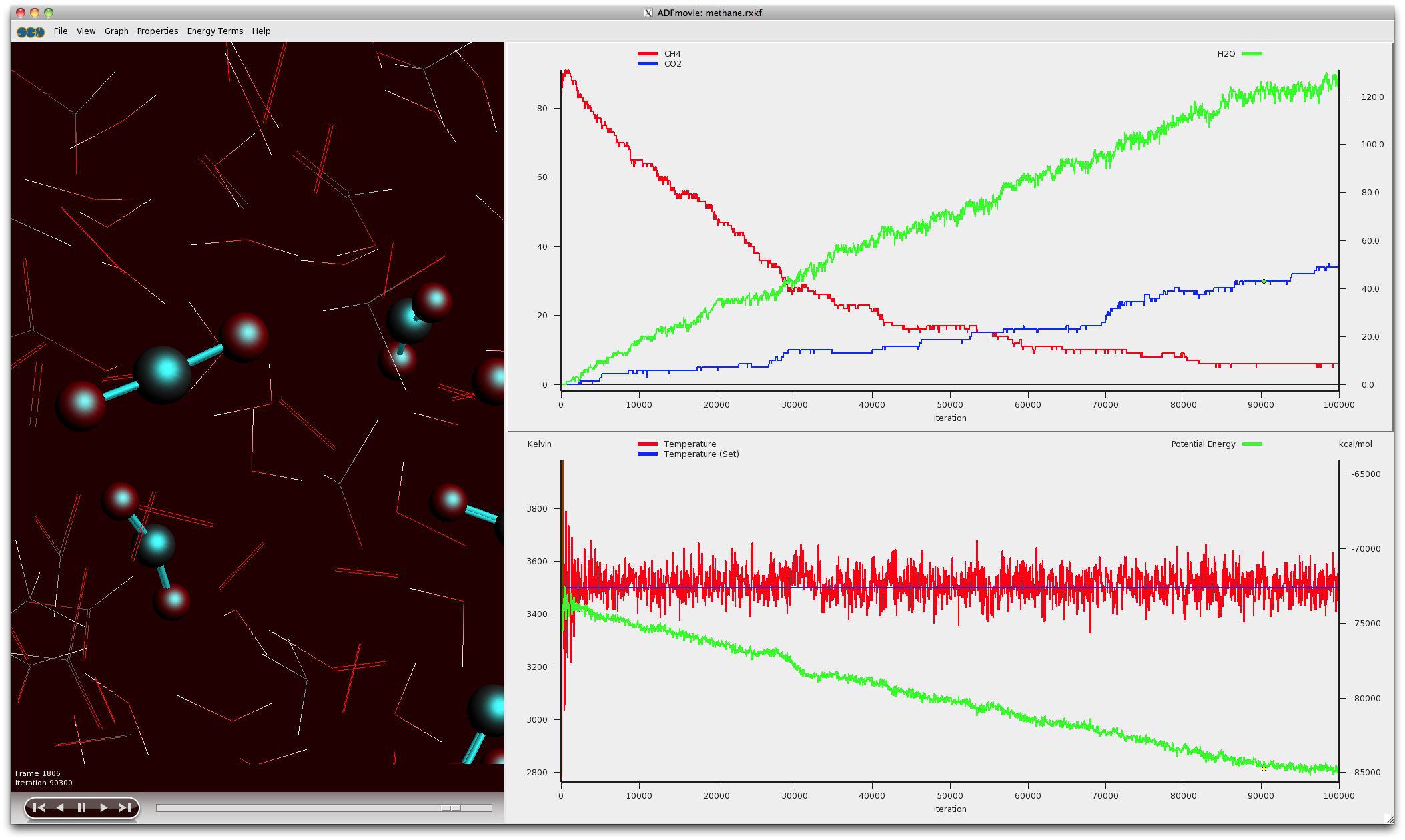
A chemical reaction being modeled with the ReaxFF program
With ADF structure, reactivity and spectroscopy of molecules is modeled with accurate and efficient DFT methods, including the latest xc functionals, relativistic effects and all-electron basis sets. The powerful molecular DFT code ADF is particularly strong in molecular properties and inorganic chemistry.
The periodic structure program BAND
Periodic structures (polymers, surfaces and crystals) can be modeled with the BAND program, featuring relativistic methods, modern xc functionals and many optical and spectroscopic properties. BAND shares a lot of functionality with ADF. Slabs are treated as real 2D systems, and nanotubes with proper 1D periodicity.
The approximate DFT programs DFTB and MOPAC
DFTB is a fast and effective Density Functional based approach to model
molecules, crystals, polymers, and surfaces. It can be used as a pre-optimizer
or to get quick insight in spectra and dynamical behavior.
MOPAC is a fast approximate method to study large molecules and big periodic systems and integrates the GUI.
ReaxFF for modeling chemical reactions
ReaxFF is available with use of the powerful GUI to run and analyze reactive molecular dynamics simulations based upon the reactive force field method of van Duin and coworkers.
COSMO-RS for fluid thermodynamics
With the COSMO-RS implementation including a database of over 1800 compounds many thermodynamic properties of solutions and mixtures can be predicted.
Not all product information on our website is available in English and German, but our sales experts are happy to assist you in either language. Please schedule a consulation appointment via email at