
")















Übersicht über Software aus dem Chemie- und Life-Science-Bereich
Die von ADDITIVE vertriebenen Chemie-Softwareprodukte decken eine große Bandbreite an Einsatzmöglichkeiten ab – angefangen bei Chemiesoftware, die Lehrende bei der verständlichen Aufbereitung ihres Unterrichtsmaterials für chemische Grundlagen unterstützt, über Softwarepakete zum Erstellen von Abbildungen und Bestimmen einfacher chemischer/physikalischer Eigenschaften bis hin zu molekularer Modellierung und Software für quantenchemische Berechnungen.
Die folgende Liste gibt einen schnellen Überblick über die ADDITIVE-Palette an Chemiesoftware. Bei Fragen und für weitere Informationen steht unser Chemie-Team für Sie per E-Mail an
Legende möglicher Funktionen
Amsterdam Modeling Suite - AMS/Scientific Computing-Produktfamilie
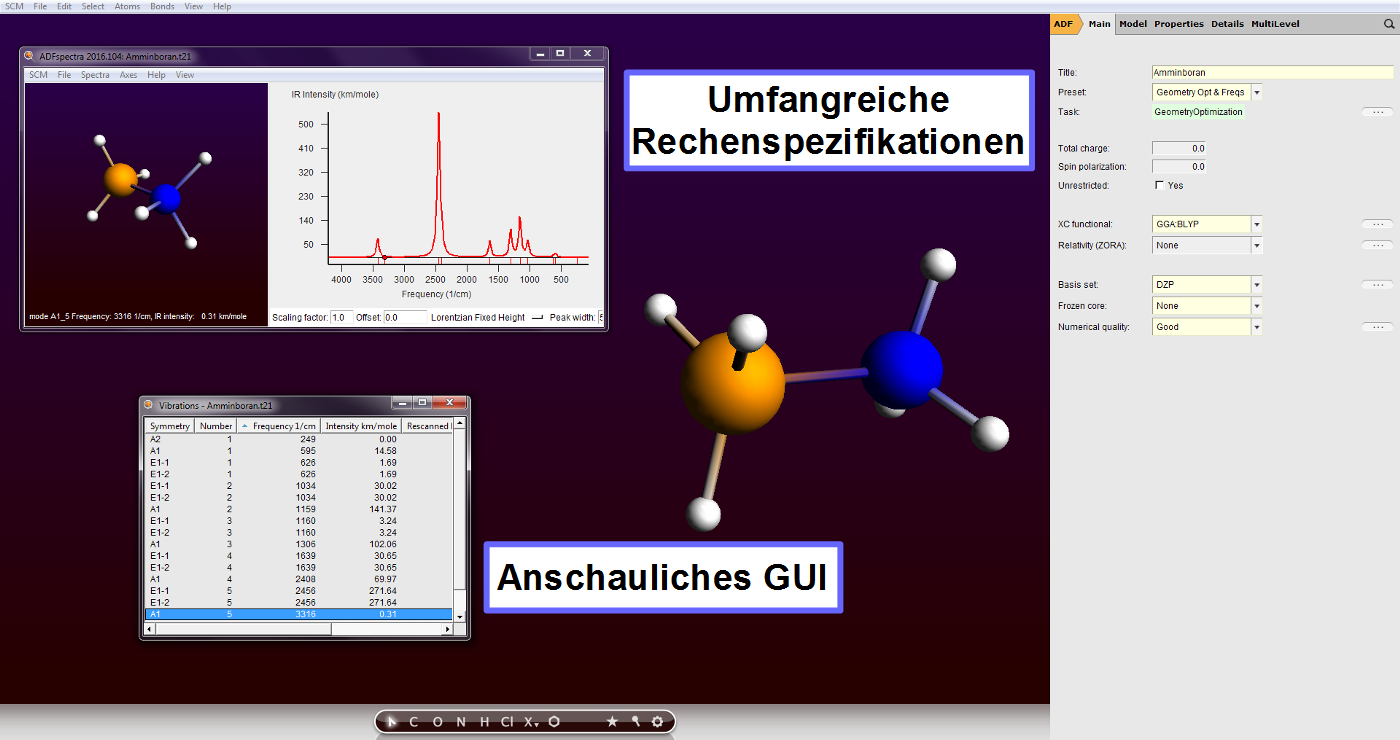
Die ADF Modeling Suite ist ein modular aufgebautes Softwarepaket zur Modellierung chemischer und physikalischer Eigenschaften. Es besteht aus den Anwendungen:
- ADF zur molekularen Modellierung
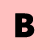
 (inkl. GUI
(inkl. GUI  )
) - BAND zur Modellierung periodischer Strukturen (inkl. GUI )
- DFTB zur Modellierung von Molekülen, Kristallen, Polymeren und Oberflächen nach dem Dichtefunktional-basierten Ansatz (inkl. GUI )
- ReaxFF zur Modellierung chemischer Reaktionen
 (inkl. GUI )
(inkl. GUI ) - COSMO-RS zur Vorhersage von thermodynamischen Eigenschaften von Lösungen und Mixturen
 (inkl. GUI )
(inkl. GUI )
Revvity Signals Software-Produktfamilie
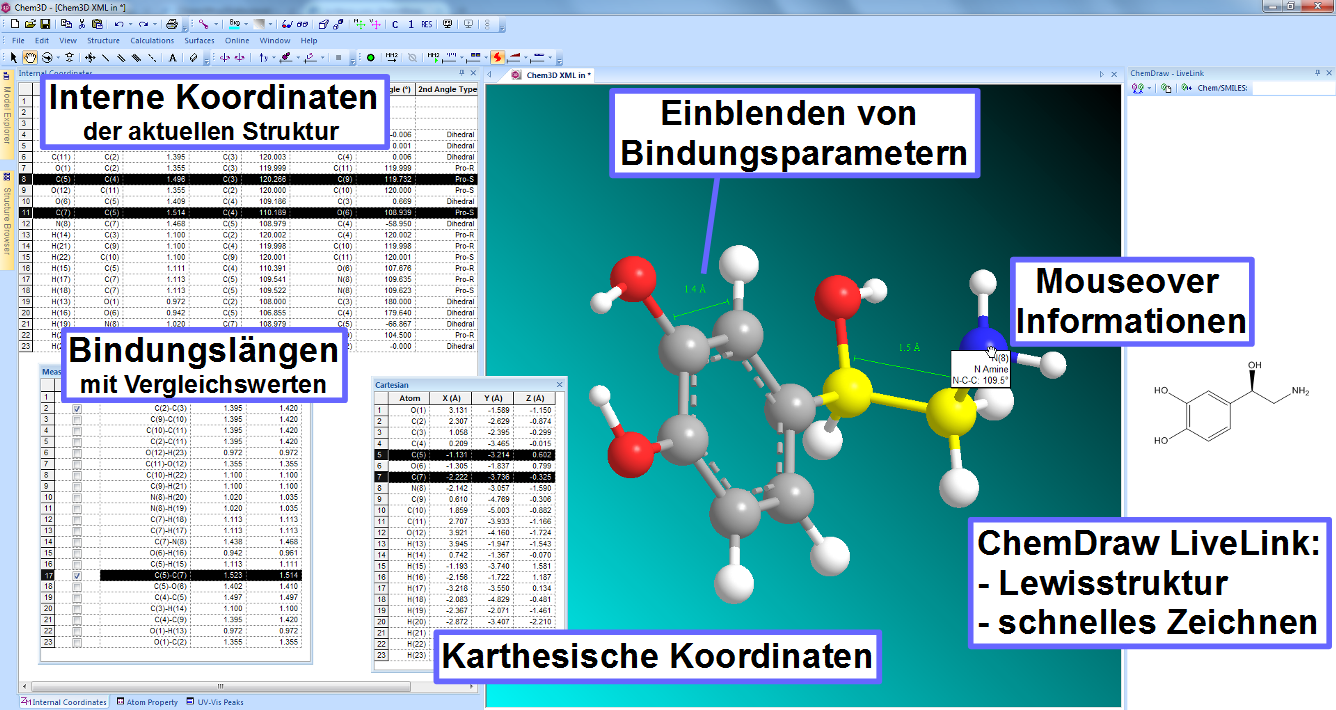
Die Revvity Signals Software-Produktfamilie besteht aus zwei verschiedenen Programmpaketen, ChemDraw und Signals ChemDraw, die jeweils mehrere verschiedene Applikationen zum chemischen und biologischen Zeichnen, zur Datenbankabfrage sowie zur Visualisierung und zur Analyse chemischer und biologischer Daten enthalten.
- ChemDraw
Intelligentes Programm zum schnellen und einfachen Erstellen von publikationsreifen Abbildungen sowie zum Untersuchen strukturierter Aufgaben im chemischen sowie biologischen Bereich - Signals ChemDraw

Alle Features, die auch ChemDraw bietet, jedoch mit erweiterten 3D-Fähigkeiten und Schnittstelleneigenschaften zu Drittanbietersoftware - Signals Notebook
Leistungsstarkes webbasiertes, elektronisches Laborjournal zum lückenlosen Erfassen und Organisieren der Laborexperimente und deren Ergebnisse in einem intuitiven Workflow zur wissenschaftlichen Zusammenarbeit - TIBCO Spotfire
Eine der führenden Enterprise Analyse- und Datenerforschungsplattformen mit extrem leistungsstarken Algorithmen, weitreichender Skalierbarkeit und hoher Datensicherheit
GAUSSIAN-Produktfamilie
Die GAUSSIAN-Produktfamilie bietet Lösungen für quantenchemische Berechnungen in der Chemie.
- GAUSSIAN
Quantenchemische Berechnungen sowie Molecular Modelling mit einer Vielzahl an Methoden und Basissätzen sowie der Möglichkeit diese benutzerdefiniert anzupassen - GAUSS View als grafische Benutzeroberfläche zu GAUSSIAN
Erstellen und Visualisieren von Molekülen und Jobs sowie ansprechende Darstellung der Ergebnisse